
 成为VIP
成为VIP
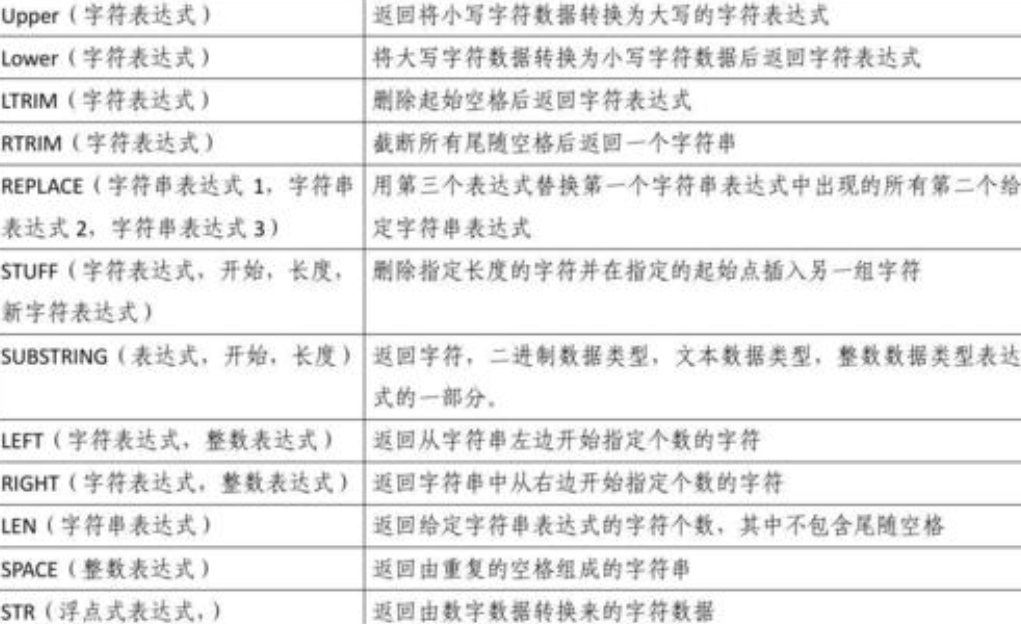
使用stringr处理字符串
正则表达式(regular expression,regexp)是处理字符串的核心步骤。正则表达式的用处非常大,字符串通常包含的是非结构化或半结构化数据shell判断字符串相等,正则表达式可以用简练的语言来描述字符串中的模式。
stringr包函数处理脏数据可谓是屡试不爽,例如:网络爬取的数据、平台反馈数据、用户数据、医疗数据
1、最基础的就是字符串拼接函数:str_c,str_join
2、数据匹配函数:inner_join、right_join、left_join、semi_join、anti_join等,其实这个就和sql中的一样。在你匹配数据的时候经常出现匹配误差很大,而实际上很多数据本身是匹配的,那么你赶紧用用str_trim去除数据中的空格,还有数据的格式,这个就很容易从报错中知道。
3、字符串的计数str_count: 字符串计数、str_length: 字符串长度,至于怎么用今天就不写了。
4、数据拆分函数:str_split、str_split_fixed,两者的 区别在于输出数据格式不同、参数可选不同
5、匹配、筛选、清理对应格式数据,str_replace、str_replace_all,两者主要在于前者只有匹配第一个与之相对应格式的数据,而后者会处理所有,函数中均有三个参数:数据列column或者文本数据等,匹配模式pattern(可根据需求使用固定格式的匹配方式、还可用正则表达式编写相应的通用pattern),替换的值
6、定位相应格式数据的位置:str_locate、str_locate,这个就比较方便,可以定位到你想要知道的数据具体位置进而去做处理
7、检查匹配字符串的字符函数:str_detect,这个函数会判断相应pattern的字符,它并不是判断两个值是否相等,而是去具体的值中搜索是否存在相应模式的值shell判断字符串相等,然后返回TRUE/FALSE
8、从字符串中提取匹配组:str_match、str_match_all,这个函数就与前面的str_replace不同,它是匹配你想要的pattern值返回给你,str_replace是清除。其实从字符串中提取匹配字符函数str_extract与它有点类似
————————————————版权声明:本文为CSDN博主「LEEBELOVED」的原创文章,遵循 CC 4.0 BY-SA 版权协议,转载请附上原文出处链接及本声明。原文链接:
导入包
library(tidyverse)
library(stringr)为了将这个功能运用与我们的微生物领域,我在这里导入一一份扩增子数据
library(phyloseq)
#导入otu表格
otu = read.delim("./otutab.txt",row.names = 1)
head(otu)
otu = as.matrix(otu)
str(otu)
#导入注释文件
tax = read.delim("./taxonomy.txt",row.names = 1)
head(tax)
tax = as.matrix(tax)
# taxa_names(tax)
#导入分组文件
map = read.delim("./metadata.tsv",row.names = 1)
head(map)
# #导入进化树
# tree = read.tree("./otus.tree")
# tree
ps <- phyloseq(otu_table(otu, taxa_are_rows=TRUE),
sample_data(map) ,
tax_table(tax)
# phy_tree(tree)
)
ps
vegan_otu <- function(physeq){
OTU <- otu_table(physeq)
if(taxa_are_rows(OTU)){
OTU <- t(OTU)
}
return(as(OTU,"matrix"))
}
otu = as.data.frame(t(vegan_otu(ps)))创建字符串:如果想要在字符串中包含一个单引号或双引号,可以使用 对其进行“转义”,但是这意味着,如果想要在字符串中包含一个反斜杠,就需要使用两个反斜杠:。最常用的是换行符n 和制表符t,你可以使用?’”‘ 或?”‘“ 调出帮助文件来查看完整的特殊字符列表。有时你还会看到”u00b5” 这样的字符串,这是一种在所有平台上都有效的非英文字符的写法:
string1 <- "This is a string"
string2 <- 'To put a "quote" inside a string, use single quotes'str_length:用于查看字符串的长度
str_c:字符串组合函数,类似的我们可以使用paste来组合字符串。
str_c("x", "y")
#> [1] "xy"
str_c("x", "y", "z")
#> [1] "xyz"
# 可以使用sep 参数来控制字符串间的分隔方式:str_c("x", "y", sep = ", ")
缺失值
# 和多数R 函数一样,缺失值是可传染的。如果想要将它们输出为"NA",可以使用str_replace_na():x <- c("abc", NA)
str_c("|-", x, "-|")
#> [1] "|-abc-|" NA
str_c("|-", str_replace_na(x), "-|")
#> [1] "|-abc-|" "|-NA-|"
# 如以上代码所示,str_c() 函数是向量化的,它可以自动循环短向量,使得其与最长的向量具有相同的长度:str_c("prefix-", c("a", "b", "c"), "-suffix")
#> [1] "prefix-a-suffix" "prefix-b-suffix" "prefix-c-suffix"
# 长度为0 的对象会被无声无息地丢弃。这与if 结合起来特别有用:name <- "Hadley"
time_of_day <- "morning"
birthday <- FALSE
#输出生日快乐
birthday <- TRUE
str_c(
"Good ", time_of_day, " ", name,
if (birthday) " and HAPPY BIRTHDAY",
"."
)
#> [1] "Good morning Hadley."if()函数的及简运行方式
#if判断及简形式,直接在if()后面加上TRUE的输出即可,如果需要运行很多内容可以在{}中添加。if (TRUE) " and HAPPY BIRTHDAY"
if (F) 5向量合并为字符串
要想将字符向量合并为字符串,很有用的功能:
# 要想将字符向量合并为字符串,可以使用collapse() 函数:str_c(c("x", "y", "z"), collapse = ", ")
#> [1] "x, y, z"微生物注释文件修改
扩增子注释文件按照不同级别分为六到七列,我们Qiime1的输出文件却只有一列,在这里我们这样操作重现:
#
head(tax)
# 仿照Qiime1的分隔符;str_c(tax[1,], collapse = " ;")
字符串取子集
# 可以使用str_sub() 函数来提取字符串的一部分。除了字符串参数外,str_sub() 函数中还有start 和end 参数,它们给出了子串的位置(包括start 和end 在内):x = tax[1,]
x = paste("P__",x[2],sep = "")
#Qiime1注释的结果中是下面的格式,如果我们要去除前面的 P__,直接简单粗暴设定开始提取第四个字符以后的字符即可。即使字符串过短,str_sub() 函数也不会出错,它将返回尽可能多的字符:x
str_sub(x, 4)
# 负数表示从后往前数
str_sub(x, -3, -1)
#> [1] "ple" "ana" "ear"
# 注意,即使字符串过短,str_sub() 函数也不会出错,它将返回尽可能多的字符:str_sub("a", 1, 5)
#> [1] "a"字符串开头大小写的转换迅速将微生物注释开头大写去除
x = tax[1,]
x
# 还可以使用str_sub() 函数的赋值形式来修改字符串:str_to_lower() 函数将文本转换为小写
str_sub(x, 1, 1) <- str_to_lower(str_sub(x, 1, 1))
x
#> [1] "apple" "banana" "pear"语言应用于排序
受区域设置影响的另一种重要操作是排序。R 基础包中的order() 和sort() 函数使用当前区域设置对字符串进行排序。如果需要更强大的、可以在不同计算机间使用的排序操作,那么可以使用str_sort() 和str_order() 函数,它们可以使用locale 参数来进行区域设置:
x <- c("apple", "eggplant", "banana")
str_sort(x, locale = "en") # 英语
#> [1] "apple" "banana" "eggplant"
str_sort(x, locale = "haw") # 夏威夷语
#> [1] "apple" "eggplant" "banana"使用正则表达式进行模式匹配
通过str_view() 和str_view_all() 函数来学习正则表达式。这两个函数接受一个字符向量和一个正则表达式,并显示出它们是如何匹配的。我们先从非常简单的正则表达式开始,然后循序渐进地学习更加复杂的正则表达式。一旦掌握了模式匹配,你就知道如何将这种思想应用于不同的stringr 函数了。
这个怎么没有输出呢个?怎么回事?
# 最简单的模式是精确匹配字符串:x <- c("apple", "banana", "pear")
x
a = str_view(x, "an")
?str_view
# 另一个更复杂一些的模式是使用.,它可以匹配任意字符(除了换行符):str_view(x, ".a.")
x <- c("apple", "banana", "pear")
str_view(x, "^a")
stringr::str_view(x, "a$")
x <- "1888 is the longest year in Roman numerals: MDCCCLXXXVIII"
str_view(x, "CC?")判断字符是否在向量中存在
要想确定一个字符向量能否匹配一种模式,可以使用str_detect() 函数。它返回一个与输入向量具有同样长度的逻辑向量:
x <- c("apple", "banana", "pear")
str_detect(x, "e")
#> [1] TRUE FALSE TRUE记住,从数学意义上来说,逻辑向量中的FALSE 为0,TRUE 为1。这使得在匹配特别大的向量时,sum() 和mean() 函数能够发挥更大的作用:这些作用会很有用:例如我们在微生物表格中想知道注释为某一个分类的OTU到底有多少个?
# 有多少个以t开头的常用单词?sum(str_detect(words, "^t"))
#> [1] 65
# 以元音字母结尾的常用单词的比例是多少?mean(str_detect(words, "[aeiou]$"))
#> [1] 0.277
x = tax[,2]
x
#查看Actinobacteria门一共注释了662个OTU。sum(str_detect(x, "Actinobacteria"))当逻辑条件非常复杂时(例如,匹配a 或b,但不匹配c,除非d 成立),一般来说,相对于创建单个正则表达式,使用逻辑运算符将多个str_detect() 调用组合起来会更容易。例如,从注释文件中统计不属于Actinobacteria的OTU数量。其次我们经常需要对数据库或者矩阵进行操作,这里使用filter即可。当然格式需要转化为tibble。
sum(!str_detect(x, "Actinobacteria"))
tax = as.data.frame(tax)
head(tax)
tax = as.tibble(tax)
Stax = tax %>%
filter(str_detect(Phylum, "Actinobacteria"))
dim(Stax)str_detect() 函数的一种变体是str_count(),后者不是简单地返回是或否,而是返回字符串中匹配的数量:
x <- c("apple", "banana", "pear")
str_count(x, "a")
#> [1] 1 3 1提取匹配内容
要想提取匹配的实际文本,我们可以使用str_extract() 函数。为了说明这个函数的用法,我们需要一个更加复杂的示例。
stringr::sentences
length(sentences)
#> [1] 720
head(sentences)假设我们想要找出包含一种颜色的所有句子。首先,我们需要创建一个颜色名称向量,然后将其转换成一个正则表达式:现在我们可以选取出包含一种颜色的句子,再从中提取出颜色,就可以知道有哪些颜色了:
这个函数很重要,这是我们进行爬虫晋经常需要做的。
colors <- c(
"red", "orange", "yellow", "green", "blue", "purple"
)
color_match <- str_c(colors, collapse = "|")
color_match
#> [1] "red|orange|yellow|green|blue|purple"
# 现在我们可以选取出包含一种颜色的句子,再从中提取出颜色,就可以知道有哪些颜色了:has_color <- str_subset(sentences, color_match)
has_color
matches <- str_extract(has_color, color_match)
head(matches)
#> [1] "blue" "blue" "red" "red" "red" "blue"提取具有多个匹配的句子。
more 1]
str_view_all(more, color_match)
str_extract(more, color_match)# 这是stringr 函数的一种通用模式,因为单个匹配可以使用更简单的数据结构。要想得到所
# 有匹配,可以使用str_extract_all() 函数,它会返回一个列表:str_extract_all(more, color_match)
#> [[1]]
#> [1] "blue" "red"
#>
#> [[2]]
#> [1] "green" "red"
#>
#> [[3]]
#> [1] "orange" "red"
# 如果设置了simplify = TRUE,那么str_extract_all() 会返回一个矩阵,其中较短的匹配
# 会扩展到与最长的匹配具有同样的长度:str_extract_all(more, color_match, simplify = TRUE)
#> [,1] [,2]
#> [1,] "blue" "red"
#> [2,] "green" "red"
#> [3,] "orange" "red"
x <- c("a", "a b", "a b c")
str_extract_all(x, "[a-z]", simplify = TRUE)
#> [,1] [,2] [,3]
#> [1,] "a" "" ""
#> [2,] "a" "b" ""
#> [3,] "a" "b" "c"str_extract_all:这个函数是在stringr包下面的一个函数,在做数据清洗的时候还是很有用的,大概用法就是去提取一个字符串下的某种内容,按照一些自己想要的规则,具体用法如下:
x<-"abacdef12g"
str_extract_all(x,"[f0-9]")限时特惠:本站每日持续更新海量展厅资源,一年会员只需29.9元,全站资源免费下载
站长微信:zhanting688


{kind=link}